Research
We explore the interfaces of recently-emerging synthetic and processing technologies in organic synthesis. Our goals are to:
1) unearth new chemistry for formation of Carbon-Carbon or Carbon-Heteroatom bonds
2) tame high-energy, reactive intermediates for use in oxidation or reduction reactions
3) develop sustainable, safer and scalable methods as alternatives for existing chemical transformations.
Synthetic Photoelectrochemistry

Single Electron Transfer (SET) chemistry affords unconventional reactivity modes & intermediates, serving as a platform for innovative bond constructions, deconstructions and functional group transformations. SET chemistry can be easily accessed by two key vehicles, namely: 1) visible light photoredox catalysis (PRC) and 2) synthetic organic electrochemistry (SOE). Both methods offer phenomenal capabilities to chemists & have changed the landscape of small molecule synthesis. However, both methods suffer fundamental drawbacks. In visible light PRC, the available energy for SET processes is constrained by visible photon energy. Sacrificial oxidants/reductants are needed for net-oxidative/reductive processes, which are atom uneconomical and interfere with downstream chemistry. In SOE, high applied potentials can often encourage unselective, deleterious redox processes.
PRC and SOE are often regarded as competing technologies. Their fusion has been largely overlooked, but overcomes multiple limitations of the parent technologies.
The Barham Lab harnesses the synergy of PRC and SOE: "Synthetic Photoelectrochemistry" (PEC) in small molecule organic synthesis.
1) electrochemically-mediated PhotoRedox Catalysis - "e-PRC"
Electrochemistry is directly involved in the PRC cycle. This concept has three benefits:
(i) Electrochemistry builds up a base layer of redox energy, 'topped-up' by photon redox energy, thereby generating "super-redox" agents in a transient fashion. Substrates can be engaged beyond the reach of PRC and SOE, selectively under mild conditions.
(ii) Sacrificial reductants/oxidants in PRC are replaced by cathodic/anodic current, increasing atom economy & removing interfering by-products.
(iii) electrogenerated radical ion photocatalysts (PCs) are colored, but neutral precursors need not be. This expands the scope of molecular architechtures that can be used as photocatalysts.
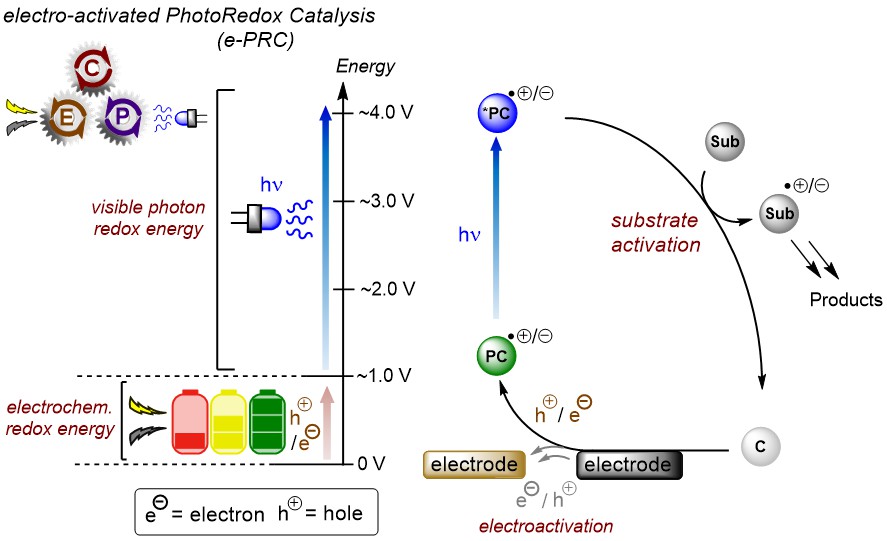
The mechanism isn't as simple as drawn above. Radical ion photocatalysts typically live shorter than diffusion, but function successfully in catalysis. Moreover, their photochemistry occurs at higher excited states than the first excited state, breaking Kasha's rule.
Substrate-radical ion preassembly is necessary for reactivity, allowing ultrashort-lived excited states to participate in SET and allowing photophysical deactivations to be circumvented.
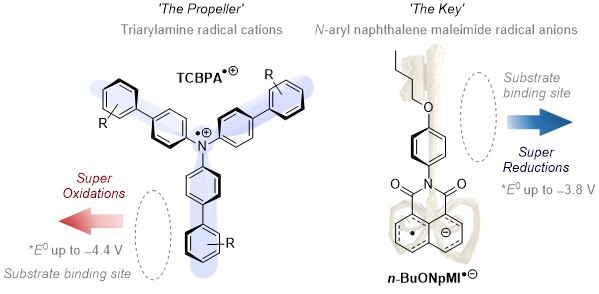
AK Barham has developed radical ion photocatalysts for both super-oxidations (up to ~ +4.4 V vs. SCE) and super-reductions (up to ~ -3.8 V vs. SCE). These were used to engage highly electron deficient arenes in oxidation, or activated alcohols in reduction, ultimately forming or cleaving strong carbon-heteroatom (C-N, C-O) bonds.
See Publications for further information.
2) interfacial PhotoElectroChemistry - "dPEC"
Photon energy is harnessed by a "photoelectrode" which engages substrates at the electrode surface in a mild fashion, offsetting high applied potentials otherwise required in the absence of light. Mirrors the concept of a photoelectrochemical cell, but for use with organic substrates instead of oxidation of H2O:

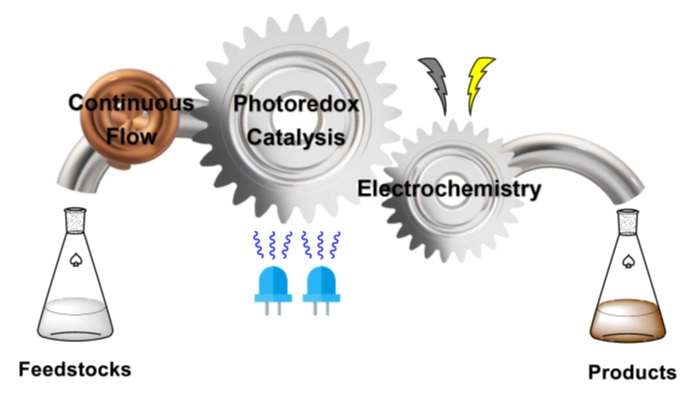
Flow Chemistry
1) Heterogeneous flow photochemistry
Photochemical reactions suffer on scale-up due to the physical constraints governing transfer of photons to (or from) the reaction. Processing in continuous flow (CF) offers short path lengths for light transmission that markedly enhance reaction efficiency. The application of backpressure on the flow allows us to safely use gaseous reagents and to handle reactions that evolve gaseous products.

Slurry-capable / liquid-liquid flow reactor


Gas-liquid flow reactor
For example research projects, check out this webinar. (from 47:22 min)
Check out our latest publication in Green Chem., handling diazo compounds in flow (without the bang!) in a catalyst-free photocyclopropanation of heterocycles under safe, sustainable and scalable conditions.
The Barham Lab leverages CF for enabling and scaling photochemical, electrochemical and photoelectrochemical reactions.
2) Microwave flow chemistry
***Coming Soon***
See Publications for further information.
Joshua Philip Barham - 05.12.2023 12:30

