

![]()
Elusive Transmetalation Intermediate in Copper-Catalyzed Conjugate Additions: Direct NMR Detection of an Ethyl Group Attached to a Binuclear Phosphoramidite Copper Complex
F. Rekowski, C. Koch,R. M. Gschwind, J. Am. Chem. Soc., 2014, 136 (32), 11389-11395.
Copper-catalyzed asymmetric conjugate addition reactions are a very powerful and widely applied method for enantioselective carbon–carbon bond formation. However, structural and mechanistic insight into these famous reactions has been very limited so far. In this article, the first direct experimental detection of transmetalation intermediates in copper-catalyzed reactions is presented. Special combinations of 1H,31P HMBC spectra allow for the identification of complexes with chemical bonds between the alkyl groups and the copper complexes. For the structural characterization of these transmetalation intermediates, a special approach is applied, in which samples using enantiopure ligands are compared with samples using enantiomeric mixtures of ligands. It is experimentally proven, for the first time, that the dimeric copper complex structure is retained upon transmetalation, providing an intermediate with mixed trigonal/tetrahedral coordination on the copper atoms. In addition, monomeric intermediates with one ligand, but no intermediates with two ligands, are detected. These experimental results, in combination with the well-known optimal ligand-to-copper ratio of 2:1 in synthetic applications, allow us to propose that a binuclear transmetalation intermediate is the reactive species in copper-catalyzed asymmetric conjugate addition reactions. This first direct experimental insight into the structure of the transmetalation intermediate is expected to support the mechanistic and theoretical understanding of this important class of reactions and to enable their further synthetic development. In addition, the special NMR approach presented here for the identification and characterization of intermediates below the detection limit of 1H NMR spectra can be applied also to other classes of catalyses.
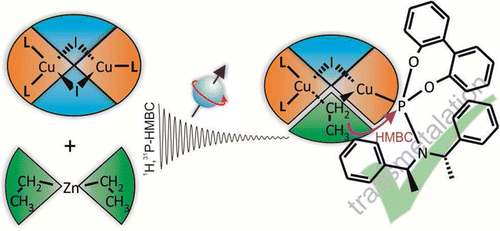
__________________________________________________________________________________________________
Elongated Gilman Cuprates: The Key to Different Reactivities of Cyano- and Iodocuprates
M. NeumeierR. M. Gschwind, J. Am. Chem. Soc., 2014, 136 (15), 5765-5772.
In the past the long-standing and very controversial discussion about a special reactivity of cyano- versus iodocuprates concentrated on the existence of higher-order cuprate structures. Later on numerous structural investigations proved the structural equivalence of iodo and cyano Gilman cuprates and their subsequential intermediates. For dimethylcuprates similar reactivities were also shown. However, the reports about higher reactivities of cyanocuprates survived obstinately in many synthetic working groups. In this study we present an alternative structural difference between cyano- and iodocuprates, which is in agreement with the results of both sides. The key is the potential incorporation of alkyl copper in iodo but not in cyano Gilman cuprates during the reaction. In the example of cuprates with a highly soluble substituent (R = Me3SiCH2) we show that in the case of iodocuprates during the reaction several copper-rich complexes are formed, which consume additional iodocuprate and provide lower reactivities. To confirm this, a variety of highly soluble copper-rich complexes were synthesized, and their molecular formulas, the position of the equilibriums, their monomers and their aggregation trends were investigated by NMR spectroscopic methods revealing extended iodo Gilman cuprates. In addition, the effect of these copper-rich complexes on the yields of cross-coupling reactions with an alkyl halide was tested, resulting in reduced yields for iodocuprates. Thus, this study gives an explanation for the thus far confusing results of both similar and different reactivities of cyano- and iodocuprates. In the case of small substituents the produced alkyl copper precipitates and similar reactivities are observed. However, iodocuprates with large substituents are able to incorporate alkyl copper units. The resulting copper-rich species have less polarized alkyl groups, i.e. gradually reduced reactivities.
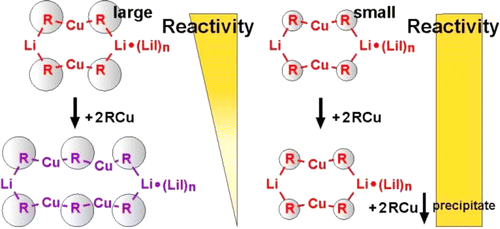
__________________________________________________________________________________________________
Structures and Interligand Interaction Pattern of Phosphoramidite Pd Complexes by NMR Spectroscopy: Modulations in Extended Interaction Surfaces as Stereoselection Mode of a Privileged Class of Ligands
During the last decade, phosphoramidites have been established as a so?called privileged class of ligands in various transition metal catalyses. However, the interactions responsible for their favorable properties have hitherto remained elusive. To address this issue, the formation trends, structural features, and interligand interaction patterns of several trans- and cis-[PdLL′Cl2] complexes have been investigated by NMR spectroscopy. The energetic contribution of their interligand interactions has been measured experimentally using the supramolecular balance for transition?metal complexes. The resulting energetics combined with an analysis of the electrostatic potential surfaces reveal that in phosphoramidites not only the aryl groups but the complete (CH)CH3Ph moieties of the amine side chains form extended quasi-planar CH?π and π?π interaction surfaces. Application of the supramolecular balance has shown that modulations in these extended interaction surfaces cause energetic differences that are relevant to enantioselective catalysis. In addition, the energetics of these interligand interactions are quite independent of the actual structures of the complexes. This is shown by similar formation and aggregation trends of complexes with the same ligand but different structures. The extended quasi?planar electrostatic interaction surface of the (CH)CH3Ph moiety explains the known pattern of successful ligand modulation and the substrate specificity of phosphoramidites. Thus, we propose modulations in these extended CH-π and π-π interaction areas as a refined stereoselection mode for these ligands. Based on the example of phosphoramidites, this study reveals three general features potentially applicable to various ligands in asymmetric catalysis. First, specific combinations of alkyl and aryl moieties can be used to create extended anisotropic interaction areas. Second, modulations in these interaction surfaces cause energetic differences that are relevant to catalytic applications. Third, bulky substituents with matching complementary interaction surfaces should not only be considered in terms of steric hindrance but also in terms of attractive and repulsive interactions, a feature that may often be underestimated in asymmetric catalysis.

__________________________________________________________________________________________________
Ligand exchange reactions in Cu(III) complexes: mechanistic insights by combined NMR and DFT studies
M. Neumeier,
NMR studies of 13C/12C isotopic patterns in Cu(III) intermediates and reaction products together with DFT calculations of possible reaction pathways indicate an intermolecular SN2 like substitution mechanism for ligand exchange reactions in square planar Cu(III) complexes, which is proposed to be slow compared to reductive elimination at synthetic conditions.

__________________________________________________________________________________________________
Highly diastereoselective Csp3–Csp2 Negishi cross-coupling with 1,2-, 1,3- and 1,4-substituted cycloalkylzinc compounds
T. Thaler, B. Haag, A. Gavryushin, K. Schober, E. Hartmann, R. M. Gschwind, H. Zipse, P. Mayer, P. Knochel, Nature Chemistry, 2010, 2, 125-130.
Stereoselective functionalizations of organic molecules are of great importance to modern synthesis. A stereoselective preparation of pharmaceutically active molecules is often required to ensure the appropriate biological activity. Thereby, diastereoselective methods represent valuable tools for an efficient set-up of multiple stereocentres. In this article, highly diastereoselective Csp3 Negishi cross-couplings of various cycloalkylzinc reagents with aryl halides are reported. In all cases, the thermodynamically most-stable stereoisomer was obtained. Remarkably, this diastereoselective coupling was successful not only for 1,2-substituted cyclic systems, but also for 1,3- and 1,4-substituted cyclohexylzinc reagents. The origin of this remote stereocontrol was investigated by NMR experiments and density functional theory calculations. A detailed mechanism based on these experimental and theoretical data is proposed.
Nikola Kastner-Pustet - 02.10.2025 12:00


Phone: 0941 943-4626
Fax: 0941 943-4617
Room: CH23.1.80
E-Mail