

![]()
Direct catalytic transformation of white phosphorus into arylphosphines and phosphonium salts
U. Lennert, P. B. Arockiam, V. Streitferdt, D. J. Scott, C. Rödl, R. M. Gschwind, R. Wolf, Nature Catalysis 2019, 2, 1101-1106.
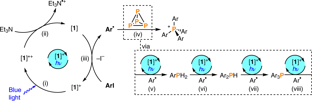
Phosphorus compounds are ubiquitous in the chemical sciences, finding applications throughout industry and academia. Of particular interest to synthetic chemists are organophosphorus compounds, which contain P–C bonds. However, state-of-the-art processes for the synthesis of these important materials rely on an inefficient, stepwise methodology involving an initial oxidation of white phosphorus (P4 organofunctionalization reactions have remained elusive, as they require multiple P–P bond-breaking and P–C bond-forming events to break down the P4. Using blue-light photocatalysis, this method directly affords valuable triarylphosphines and tetraarylphosphonium salts in a single reaction step.
__________________________________________________________________________________________________
Combination of illumination and high resolution NMR spectroscopy: Key features and practical aspects, photochemical applications, and new concepts
P. Nitschke, L. Nanjundappa, R.M. Gschwind, Prog. Nucl. Magn. Reson. Spectrosc., 2019, 114-115, 86-134.
In the last decade, photochemical and photocatalytic applications have developed into one of the dominant research fields in chemistry. However, mechanistic investigations to sustain this enormous progress are still relatively sparse and in high demand by the photochemistry community. UV/Vis spectroscopy and EPR spectroscopy have been the main spectroscopic tools to study the mechanisms of photoreactions due to their higher time resolution and sensitivity. On the other hand, application of NMR in photosystems has been mainly restricted to photo-CIDNP, since the initial photoexcitation was thought to be the single key to understand photoinduced reactions. In 2015 the Gschwind group showcased the possibility that different reaction pathways could occur from the same photoexcited state depending on the reaction conditions by using in situ LED illumination NMR. This was the starting point to push the active participation of NMR in photosystems to its full potential, including reaction profiling, structure determination of intermediates, downstream mechanistic studies, dark pathways, intermediate sequencing with CEST etc. Following this, multiple studies using in situ illumination NMR have been reported focusing on mechanistic investigations in photocatalysis, photoswitches, and polymerizations. The recent increased popularity of this technique can be attributed to the simplicity of the experimental setup and the availability of low cost, high power LEDs. Here, we review the development of experimental design, applications and new concepts of illuminated NMR. In the first part, we describe the development of different designs of NMR illumination apparatus, illuminating from the bottom/side/top/inside, and discuss their pros and cons for specific applications. Furthermore, we address LASERs and LEDs as different light sources as well as special cases such as UVNMR(-illumination), FlowNMR, NMR on a Chip etc. To complete the discussion on experimental apparatus, the advantages and disadvantages of in situ LED illumination NMR versus ex situ illumination NMR are described. The second part of this review discusses different facets of applications of inside illumination experiments. It highlights newly revealed mechanistic and structural information and ideas in the fields of photocatalyis, photoswitches and photopolymerization. Finally, we present new concepts and methods based on the combination of NMR and illumination such as sensitivity enhancement, chemical pump probes, experimental access to transition state combinations and NMR actinometry. Overall this review presents NMR spectroscopy as a complementary tool to UV/Vis spectroscopy in mechanistic and structural investigations of photochemical processes. The review is presented in a way that is intended to assist the photochemistry and photocatalysis community in adopting and understanding this astonishingly powerful in situ LED illumination NMR method for their investigations on a daily basis.
__________________________________________________________________________________________________
LED-Illuminated NMR Spectroscopy: A Practical Tool for Mechanistic Studies of Photochemical Reactions
Y.Ji, D. A. DiRocco, J. Kind, C. M. Thiele, R. M. Gschwind, M. Reibarkh, ChemPhotoChem 2019, 3, 984-992.
This Concept article highlights the development of a novel analytical tool, LED-NMR (a combination of in-situ light illumination using a light?emitting diode and NMR spectroscopy) and its variant UVNMR (LED-NMR coupled with UV/Vis absorption spectroscopy), as well as their applications in the mechanistic investigation of light?induced transformations. The utility of these new tools has been demonstrated by providing rich kinetic and structural data of reaction species offering mechanistic insights into photochemical and photocatalytic reactions. Furthermore, NMR actinometry has been recently developed as a practical and simple method for quantum yield measurements. Quantum yield is an important parameter in photo-induced processes, but is rarely measured in practice because of the barriers associated with traditional actinometry. These new tools and techniques streamline measurements of the quantum efficiency while affording informative mechanistic insights into photochemical reactions. We anticipate these techniques will enable chemists to further advance the rapidly emerging photochemistry field.

__________________________________________________________________________________________________
Photoinitiated carbonyl-metathesis: deoxygenative reductive olefination of aromatic aldehydes via photoredox catalysis
S. Wang, L. Nanjundappa, J. Hioe, R. M. Gschwind, B. König, Chem. Sci., 2019, 10, 4580-4587.
Carbonyl–carbonyl olefination, known as McMurry reaction, represents a powerful strategy for the construction of olefins. However, catalytic variants that directly couple two carbonyl groups in a single reaction are less explored. Here, we report a photoredox-catalysis that uses B2pin2 as terminal reductant and oxygen trap allowing for deoxygenative olefination of aromatic aldehydes under mild conditions. This strategy provides access to a diverse range of symmetrical and unsymmetrical alkenes with moderate to high yield (up to 83%) and functional-group tolerance. To follow the reaction pathway, a series of experiments were conducted including radical inhibition, deuterium labelling, fluorescence quenching and cyclic voltammetry. Furthermore, NMR studies and DFT calculations were combined to detect and analyze three active intermediates: a cyclic three-membered anionic species, an α-oxyboryl carbanion and a 1,1-benzyldiboronate ester. Based on these results, we propose a mechanism for the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond generation involving a sequential radical borylation, “bora-Brook” rearrangement, B2pin2-mediated deoxygenation and a boron-Wittig process.
C bond generation involving a sequential radical borylation, “bora-Brook” rearrangement, B2pin2-mediated deoxygenation and a boron-Wittig process.

__________________________________________________________________________________________________
Visible Light Mediated Liberation and in situ Conversion of Fluorophosgene
D. Petzold, P. Nitschke, F. Brandl, V. Scheidler, B. Dick, R.M. Gschwind, B. König, Chem. Eur. J., 2019, 25, 361-366.
The first example for the photocatalytic generation of a highly electrophilic intermediate that is not based on radical reactivity is reported. The single electron reduction of bench stable and commercially available 4-(trifluoromethoxy)benzonitrile by an organic photosensitizer leads to its fragmentation into fluorophosgene and benzonitrile. The in situ generated fluorophosgene was used for the preparation of carbonates, carbamates, and urea derivatives in moderate to excellent yields via an intramolecular cyclization reaction. Transient spectroscopic investigations suggest the formation of a catalyst charge transfer complex dimer as the catalytic active species. Fluorophosgene as a highly reactive intermediate, was indirectly detected via its next downstream carbonyl fluoride intermediate by NMR. Furthermore, detailed NMR analyses provided a comprehensive reaction mechanism including a water dependent off cycle equilibrium.
__________________________________________________________________________________________________
Combined In Situ Illumination-NMR-UV/Vis Spectroscopy: A New Mechanistic Tool in Photochemistry
A. Seegerer, P. Nitschke, R.M. Gschwind, Angew. Chem. Int. Ed., 2018, 57, 7493-7497.
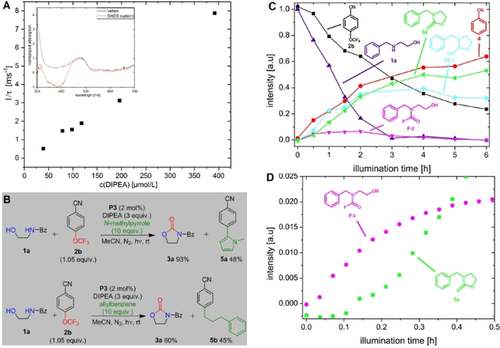
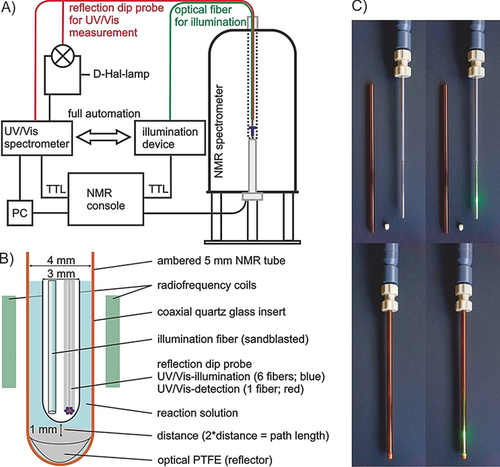
Synthetic applications in photochemistry are booming. Despite great progress in the development of new reactions, mechanistic investigations are still challenging. Therefore, we present a fully automated in situ combination of NMR spectroscopy, UV/Vis spectroscopy, and illumination to allow simultaneous and time resolved detection of paramagnetic and diamagnetic species. This optical fiber based setup enables the first acquisition of combined UV/Vis and NMR spectra in photocatalysis, as demonstrated on a conPET process. Furthermore, the broad applicability of combined UVNMR spectroscopy for light induced processes is demonstrated on a structural and quantitative analysis of a photoswitch, including rate modulation and stabilization of transient species by temperature variation. Owing to the flexibility regarding the NMR hardware, temperature, and light sources, we expect wide ranging applications of this setup in various research fields.
__________________________________________________________________________________________________
Selective Single C(sp3)–F Bond Cleavage in Trifluoromethylarenes: Merging Visible-Light Catalysis with Lewis Acid Activation
K. Chen, N. Berg, R.M. Gschwind, B. König, Am. Chem. Soc., 2017, 139, 18444-18447.
The conversion of easily available trifluoromethylarenes into aryldifluoromethyl compounds, which are valuable motifs in the pharmaceutical chemistry, is highly atom- and step-economical. However, the single C(sp3)–F bond cleavage of ArCF3 is a great challenge because of the chemical inertness of the C(sp3)–F bond and the difficult selectivity control of monodefluorination. We report here the first example of single C(sp3)–F functionalization of trifluoromethylarenes via visible-light catalysis merged with Lewis acid activation. The method allows good chemoselectivity control and shows good functional group tolerance. Mechanistic studies suggest an in situ-generated borenium cationic species as the key intermediate for C(sp3)–F bond cleavage in this reaction.

__________________________________________________________________________________________________
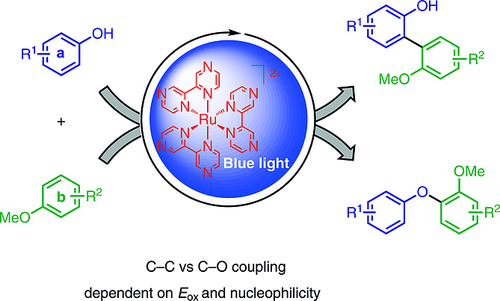
Photocatalytic Phenol–Arene C–C and C–O Cross-Dehydrogenative Coupling
Phenol-containing nonsymmetrical biaryls play an important role in natural-product synthesis, as ligands in metal catalysis, and in organic functional materials. Their synthesis through cross-coupling reactions by two-fold direct C–H activation are important and challenging transformations. In the last decade, a variety of useful oxidative methods have been developed. The key to efficiency and selectivity typically is the application of highly fluorinated solvent systems. Herein, we describe the visible-light-mediated C–C and C–O cross-coupling of electron-rich phenols and arenes by using [Ru(bpz)3](PF6)2] as photocatalyst and ammonium persulfate as terminal oxidant. The method requires no leaving group functionalities, which allows the use of simple activated arenes as starting materials. Furthermore, the approach features good chemo- and regioselectivity as well as functional group tolerance, even upon the replacement of fluoro alcohols by acetonitrile. The selectivity for the formation of nonsymmetrical biaryls is rationalized on the basis of the nucleophilicity N values and the oxidation potentials Eox of the electron?rich substrates.
__________________________________________________________________________________________________
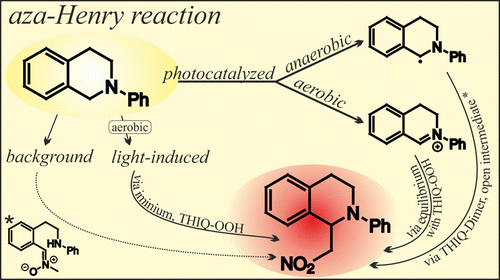
The Photocatalyzed Aza-Henry Reaction of N-Aryltetrahydroisoquinolines: Comprehensive Mechanism, H•- versus H+-Abstraction, and Background Reactions
H. Bartling, A. Eisenhofer, B. König, R.M. Gschwind, J. Am. Chem. Soc., 2016, 138 (36), 11860–11871.
The cross-dehydrogenative coupling (CDC) reaction of N-aryltetrahydroisoquinolines (THIQ) is one of the most exploited photocatalytic transformation and a test reaction for an exceptional variety of catalysts. However, its mechanism remained unclear concerning involved intermediates, reactive pathways of the amine radical cation and the influence of oxygen and the light source. Therefore, nuclear magnetic resonance (NMR), electron spin resonance (ESR) and synthetic methods were combined to provide a comprehensive picture of the reaction mechanism using Ru(bpy)3Cl2 as a photocatalyst under aerobic and anaerobic conditions. The reaction profiles and involved intermediates were monitored and analyzed by NMR spectroscopy. Several intermediates contributing to product formation were identified, the iminium ion, the hydroperoxide and dimer of THIQ, and a new ring opened intermediate, cleaved at the benzylic C–N bond. Mechanistic evidence is given that under anaerobic conditions preferentially the α-amino radical is formed by deprotonation, in contrast to the formation of iminium ions via H•-abstraction in the presence of oxygen. Further, the light-induced background reaction in the absence of the catalyst was studied in detail, revealing that the product formation rate is correlated to the intensity and wavelength of the light source and that oxygen is essential for an efficient conversion. The reaction rate and efficiency is comparable to previously reported photocatalytic systems, performed under aerobic conditions in combination with intense blue light sources. Thus, the multitude of reaction parameters investigated reveals the preference for hydrogen atom or proton abstraction in photoreactions and allows to assess the influence of experimental conditions on the mechanistic pathways.
__________________________________________________________________________________________________
Studies of a photochromic model system using NMR with ex-situ and in-situ irradiation devices
The switching behavior of a photochromic model system was investigated in detail via NMR spectroscopy in order to improve understanding of the compound itself and to provide ways to obtain insights into composition trends of a photo switchable (polymeric) material containing spiropyran/merocyanine units. In addition to the classical irradiation performed outside the magnet (ex-situ), a device for irradiation inside the NMR spectrometer (in-situ) was tested. Both setups are introduced, their advantages and disadvantages as well as their limits are described and the setup for future investigations of photochromic materials is suggested.

__________________________________________________________________________________________________
LED-Illuminated NMR Studies of Flavin-Catalyzed Photooxidations Reveal Solvent Control of the Electron-Transfer Mechanism
, R.

__________________________________________________________________________________________________
LED based NMR Illumination Device for Mechanistic Studies on Photochemical Reactions – Versatile and Simple, yet Surprisingly Powerful

__________________________________________________________________________________________________
Aggregation Effects in Visible-Light Flavin Photocatalysts: Synthesis, Structure, and Catalytic Activity of 10-Arylflavins
A series of 10-arylflavins (10-phenyl-, 10-(2′,6′-dimethylphenyl)-, 10-(2′,6′-diethylphenyl)-, 10-(2′,6′-diisopropylphenyl)-, 10-(2′-tert-butylphenyl)-, and 10-(2′,6′-dimethylphenyl)-3-methylisoalloxazine (2 a–f)) was prepared as potentially nonaggregating flavin photocatalysts. The investigation of their structures in the crystalline phase combined with 1H-DOSY NMR spectroscopic experiments in CD3CN, CD3CN/D2O (1:1), and D2O confirm the decreased ability of 10-arylflavins 2 to form aggregates relative to tetra-O-acetyl riboflavin (1). 10-Arylflavins 2 a–d do not interact by π–π interactions, which are restricted by the 10-phenyl ring oriented perpendicularly to the isoalloxazine skeleton. On the other hand, N3-H⋅⋅⋅O hydrogen bonds were detected in their crystal structures. In the structure of 10-aryl-3-methylflavin (2 f) with a substituted N3 position, weak C-H⋅⋅⋅O bonds and weak π–π interactions were found. 10-Arylflavins 2 were tested as photoredox catalysts for the aerial oxidation of 4-methoxybenzyl alcohol to the corresponding aldehyde (model reaction), thus showing higher efficiency relative to 1. The quantum yields of 4-methoxybenzyl alcohol oxidation reactions mediated by arylflavins 2 were higher by almost one order of magnitude relative to values in the presence of 1.

Nikola Kastner-Pustet - 02.10.2025 12:00


Phone: 0941 943-4626
Fax: 0941 943-4617
Room: CH23.1.80
E-Mail