

Synthesis of Natural Products and Analogues
Traceless Stereoinduction for the Enantiopure Synthesis of Substituted-2-Cyclopentenones
N. Arisetti, O. Reiser, Org. Lett. 2015, 17, 94-97
Web edition: http://dx.doi.org/10.1021/ol5032975
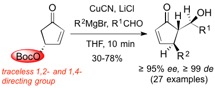
The pseudoenantiomeric 4-O-Boc- and 4-OPMP-cyclopent-2-enones, readily available from hydroxymethylenefurane on multigram scale, are demonstrated to be exceptional building blocks for the synthesis of enantiopure 4-alkyl-5-(1'-hydroxyalkyl) substituted 2-cyclopentenones and derivatives thereof. The 4-OR substituent acts as a traceless stereoinducing element, conferring not only 1,2- but also 1,4-stereocontrol with excellent selectivity. The methodology developed here was applied for the rapid synthesis of natural products and biologically active 2-cyclopentenones such as TEI-9826, guaianes and pseudoguaianolides.
Enantioselective Synthesis of Xanthatin
A. Bergmann, O. Reiser, Chem. Eur. J. 2014, 20, 7613-7615
Web edition: http://dx.doi.org/10.1002/chem.201402735
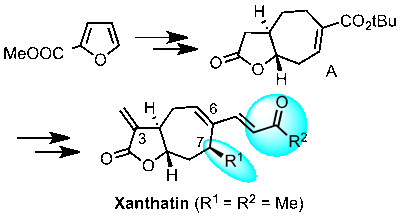
The enantioselective synthesis of the cytostatic and antibiotic Xanthatin is reported. As a key intermediate the bicyclic compound A was identified, which can be readily synthesized from 2-furoic acid in diastereo- and enantiomerically pure form. A can be functionalized regio- and stereoselectively at C-6 and C-7, allowing the facile introduction of the functionalities found in Xanthatin as well as the synthesis of derivatives thereof. Moreover, a new strategy for the introduction of the exo-methylene group at C-3, commonly found in many sesquiterpenes, was developed.
Enantioselective Synthesis of (–)-Paeonilide
K. Harrar, O. Reiser, Chem. Commun. 2012, 48, 3457-3459
Web edition: http://dx.doi.org/10.1039/C2CC18172J
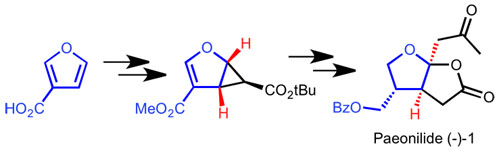
The first enantioselective synthesis of (-)-Paeonilide is reported. Starting from inexpensive furan-3-carboxylic acid the targeted monoterpene was obtained in 12 steps via an asymmetric cyclopropanation-lactonization cascade and a stereoselective side chain insertion on an acetal-like position.
First Enantioselective Total Synthesis of Arglabin
S. Kalidindi, W. B. Jeong, A. Schall, R. Bandichhor, B. Nosse, O. Reiser, Angew. Chem. 2007, 119, 6478-6481; Angew. Chem. Int. Ed. 2007, 46, 6361-6363
Web edition: http://dx.doi.org/10.1002/anie.200701584
The first enantioselective synthesis of Arglabin and its dimethylamino adduct, showing promising results in the treatment of various tumors, has been achieved.
Side-chain modified analogues of histaprodifen: Asymmetric synthesis and histamine H1-receptor activity
R. Patil,
S. Elz,
O. Reiser, Bioorg. Med. Chem. Lett. 2006, 16, 672-676
Web edition: http://dx.doi.org/10.1016/j.bmcl.2005.10.030
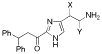
New analogues of histaprodifen with polar side chains have been stereoselectively synthesized and evaluated as histamine H1-receptor agonists. As a key transformation the asymmetric aminohydroxylation has been used, which was successfully realized for the first time on an imidazolyl derivative. While all chiral analogues proved to be weak H1-receptor antagonists, an achiral keto derivative of histaprodifen turned out to be the first 2-acylated histamine congener displaying partial H1-receptor agonism (relative potency 12%).
Enantioselective Synthesis of Furo[2,3-b]furans, a Spongiane Diterpenoid Substructure
R. Weisser,
W. Yue,
O. Reiser, Org. Lett. 2005, 7, 5353-5356
Web edition: http://dx.doi.org/10.1021/ol051457m
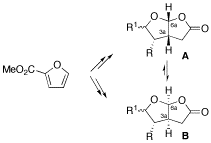
A short and enantioselective synthesis of cis-fused 5-oxo-furo[2,3-b]furans, being found in many spongiane diterpenoid natural products, is reported starting from inexpensive methyl 2-furoate. Moreover, the acid-catalyzed rearrangement of the furo[2,3-b]furan framework A to B is observed for some derivatives, suggesting a simple connection between natural products differing in the absolute configuration of the 3a,6a ring junction.
Stereoselective Synthesis of Swainsonines from Pyridines
G. Heimgärtner, D. Raatz, O. Reiser, Tetrahedron 2005, 61, 643-655
Web edition: http://dx.doi.org/10.1016/j.tet.2004.10.086
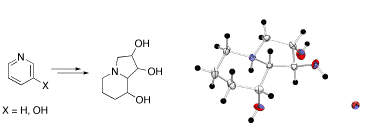
A highly efficient synthesis of swainsonine and 2,8a-di-epi-swainsonine was developed starting from readily available pyridine and 3-hydroxypyridine. In particular, it was demonstrated that the mixture of simple indolizidines, i.e. lentignosine and epi-lentiginosine, being readily available by a number of different synthetic routes, can be directly converted to swainsonine.
Facile Asymmetric Synthesis of the Core Nuclei of Xanthanolides, Guaianolides and Eudesmanolides.
B. Nosse, R. B. Chhor, W. B. Jeong, C. Böhm, O. Reiser, Org. Lett. 2003, 5, 941-944
Web edition: http://dx.doi.org/10.1021/ol034141s
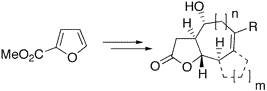
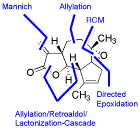
Bicyclic and tricyclic -butyrolactones with 5,7-, 5,6,5-, 5,6,6-, or 5,7,5-fused ring systems, being found in xanthanolides, eudesmanolides, and guaianolides, were readily synthesized from methyl furan-2-carboxylic acid. Key steps were a copper(I)-catalyzed asymmetric cyclopropanation, Sakurai allylations, intramolecular ene reactions, and ring-closing metathesis reactions.
Enantioselective Synthesis of Paraconic Acids.
R.B. Chhor, B. Nosse, S. Sörgel, C. Böhm, M. Seitz, O. Reiser, Chem. Eur. J. 2003, 9, 260-270
Web edition: http://dx.doi.org/10.1002/chem.200390019
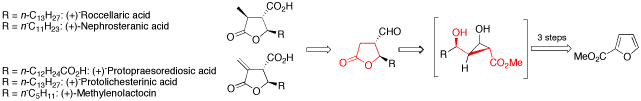
The development of a new method for the enantioselective synthesis of disubstituted -butyrolactones is reported. Based on this strategy, the total synthesis of three paraconic acids, that is (-)-roccellaric acid, (-)-nephrosteranic acid and (-)-protopraesorediosic acid, and the formal total synthesis of (-)-methylenolactocin and (-)-protolichesterinic acid is described, which are important because of their antibiotic and antitumor properties. Key steps of the synthesis are copper(I)-catalyzed asymmetric cyclopropanations of furans, highly diastereoselective Sakurai allylations, Lewis acid or Lewis base catalyzed retroaldol/lactonization cascades, and ruthenium(II)-catalyzed, intermolecular cross metathesis reactions.
Peter Kreitmeier - 07.03.2023 17:37

