

![]()
Internal Acidity Scale and Reactivity Evaluation of Chiral Phosphoric Acids with different 3,3'-Substituents in Brønsted Acid Catalysis
K. Rothermel, M. Melikian, J. Hioe, J. Greindl, J. Gramüller, M.abka, N. Sorgenfrei, T. Hausler, F. Morana. R.M. Gschwind, Chem. Sci. 2019
The concept of hydrogen bonding for enhancing substrate binding and controlling selectivity and reactivity is central in catalysis. However, the properties of these key hydrogen bonds and their catalyst-dependent variations are extremely difficult to determine directly by experiments. Here, for the first time the hydrogen bond properties of a whole series of BINOL-derived chiral phosphoric acid (CPA) catalysts in their substrate complexes with various imines were investigated to derive the influence of different 3,30-substituents on the acidity and reactivity. NMR 1H and 15N chemical shifts and 1JNH coupling constants of these hydrogen bonds were used to establish an internal acidity scale corroborated by calculations.
Deviations from calculated external acidities reveal the importance of intermolecular interactions for this key feature of CPAs. For CPAs with similarly sized binding pockets, a correlation of reactivity and hydrogen bond strengths of the catalyst was found. A catalyst with a very small binding pocket showed significantly reduced reactivities. Therefore, NMR isomerization kinetics, population and chemical shift analyses of binary and ternary complexes as well as reaction kinetics were performed to address the steps of the transfer hydrogenation influencing the overall reaction rate. The results of CPAs with different 3,30-substituents show a delicate balance between the isomerization and the ternary complex formation to be rate-determining. For CPAs with an identical acidic motif and similar sterics, reactivity and internal acidity correlated inversely. In cases where higher sterical demand within the binary complex hinders the binding of the second substrate, the correlation between acidity and reactivity breaks down.

__________________________________________________________________________________________________
Relaxation Dispersion NMR to Reveal Fast Dynamics in Brønsted Acid Catalysis: Influence of Sterics and H-Bond Strength on Conformations and Substrate Hopping
N. Lokesh, J. Hioe, J. Gramüller, R.M. Gschwind, J. Am. Chem. Soc. 2019
NMR provides both structural and dynamic information, which is key to connecting intermediates and to understanding reaction pathways. However, fast exchanging catalytic intermediates are often inaccessible by conventional NMR due its limited time resolution. Here, we show the combined application of the 1H off-resonance R1ρ NMR method and low temperature (185–175 K) to resolve intermediates exchanging on a μs time scale (ns at room temperature). The potential of the approach is demonstrated on chiral phosphoric acid (CPA) catalysts in their complexes with imines. The otherwise inaccessible exchange kinetics of the E-I ? E-II imine conformations and thermodynamic E-I:E-II imine ratios inside the catalyst pocket are experimentally determined and corroborated by calculations. The E-I ? E-II exchange rate constants (kex185 K) for different catalyst–substrate binary complexes varied between 2500 and 19?000 s–1 (τex = 500–50 μs). Theoretical analysis of these exchange rate constants revealed the involvement of an intermediary tilted conformation E-III, which structurally resembles the hydride transfer transition state. The main E-I and E-II exchange pathway is a hydrogen bond strength dependent tilting–switching–tilting mechanism via a bifurcated hydrogen bond as a transition state. The reduction in the sterics of the catalyst showed an accelerated switching process by at least an order of magnitude and enabled an additional rotational pathway. Hence, the exchange process is mainly a function of the intrinsic properties of the 3,3′-substituents of the catalyst. Overall, we believe that the present study opens a new dimension in catalysis via experimental access to structures, populations, and kinetics of catalyst–substrate complexes on the μs time scale by the 1H off-resonance R1ρ method.

__________________________________________________________________________________________________
Brønsted Acid Catalysis – The Effect of 3,3’-Substituents on the Structural Space and the Stabilization of Imine/Phosphoric Acid Complexes
Chem. Sci., 2019, 10, 5226-5234.

__________________________________________________________________________________________________
Enamine/Dienamine and Brønsted Acid Catalysis: Elusive Intermediates, Reaction Mechanisms, and Stereoinduction Modes Based on in Situ NMR Spectroscopy and Computational Studies
P. Renzi, J. Hioe, R.M. Gschwind
Over the years, the field of enantioselective organocatalysis has seen unparalleled growth in the development of novel synthetic applications with respect to mechanistic investigations. Reaction optimization appeared to be rather empirical than rational. This offset between synthetic development and mechanistic understanding was and is generally due to the difficulties in detecting reactive intermediates and the inability to experimentally evaluate transition states. Thus, the first key point for mechanistic studies is detecting elusive intermediates and characterizing them in terms of their structure, stability, formation pathways, and kinetic properties. The second key point is evaluating the importance of these intermediates and their properties in the transition state.
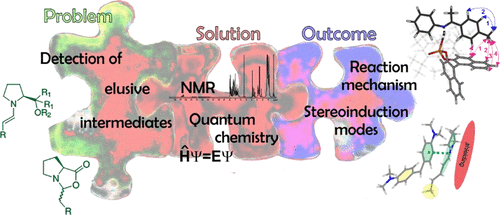
__________________________________________________________________________________________________
Decrypting Transition States by Light: Photoisomerization as a Mechanistic Tool in Brønsted Acid Catalysis
P. Renzi, J. Hioe, and R.M. Gschwind, J. Am. Chem. Soc., 2017,139, 19, 6752-6760.
Despite the wide applicability of enantioselective Brønsted acid catalysis, experimental insight into transition states is very rare, and most of the mechanistic knowledge is gained by theoretical calculations. Here, we present an alternative approach (decrypting transition state by light = DTS-hν), which enables the decryption of the transition states involved in chiral phosphoric acids catalyzed addition of nucleophiles to imines. Photoisomerization of double bonds is employed as a mechanistic tool. For this class of reactions four pathways (Type I Z, Type I E, Type II Z, Type II E) are possible, leading to different enantiomers depending on the imine configuration (E- or Z-imine) and on the nucleophilic attack site (top or bottom). We demonstrated that the imine double bond can be isomerized by light (365 nm LED) during the reaction leading to a characteristic fingerprint pattern of changes in reaction rate and enantioselectivity. This characteristic fingerprint pattern is directly correlated to the transition states involved in the transformation. Type I Z and Type II Z are demonstrated to be the competing pathways for the asymmetric transfer hydrogenation of ketimines, while in the nucleophilic addition of acetylacetone to N-Boc protected aldimines Type I E and Type II E are active. Accelerations on reaction rate up to 177% were observed for ketimines reduction. Our experimental findings are supported by quantum chemical calculations and noncovalent interaction analysis.
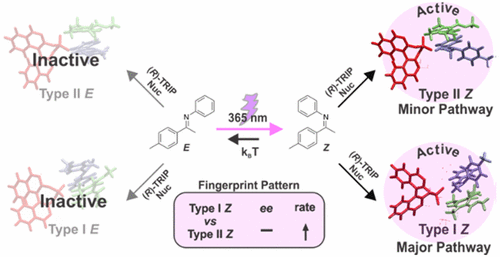
__________________________________________________________________________________________________
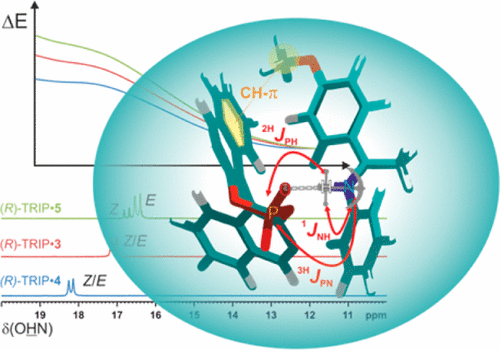
Brønsted Acid Catalysis—Structural Preferences and Mobility in Imine/Phosphoric Acid Complexes
J. Greindl, J. Hioe, N. Sorgenfrei, F. Morana, R.M. Gschwind, J. Am. Chem. Soc., 2016, 138 (49), 15965-15971.
Despite the huge success of enantioselective Brønsted acid catalysis, experimental data about structures and activation modes of substrate/catalyst complexes in solution are very rare. Here, for the first time, detailed insights into the structures of imine/Brønsted acid catalyst complexes are presented on the basis of NMR data and underpinned by theoretical calculations. The chiral Brønsted acid catalyst R-TRIP (3,3′-bis(2,4,6-triisopropylphenyl)-1,1′-binaphthyl-2,2′-diyl hydrogen phosphate) was investigated together with six aromatic imines. For each investigated system, an E-imine/R-TRIP complex and a Z-imine/R-TRIP complex were observed. Each of these complexes consists of two structures, which are in fast exchange on the NMR time scale; i.e., overall four structures were found. Both identified E-imine/R-TRIP structures feature a strong hydrogen bond but differ in the orientation of the imine relative to the catalyst. The exchange occurs by tilting the imine inside the complex and thereby switching the oxygen that constitutes the hydrogen bond. A similar situation is observed for all investigated Z-imine/R-TRIP complexes. Here, an additional exchange pathway is opened via rotation of the imine. For all investigated imine/R-TRIP complexes, the four core structures are highly preserved. Thus, these core structures are independent of electron density and substituent modulations of the aromatic imines. Overall, this study reveals that the absolute structural space of binary imine/TRIP complexes is large and the variations of the four core structures are small. The high mobility is supposed to promote reactivity, while the preservation of the core structures in conjunction with extensive π–π and CH−π interactions leads to high enantioselectivities and tolerance of different substrates.
__________________________________________________________________________________________________
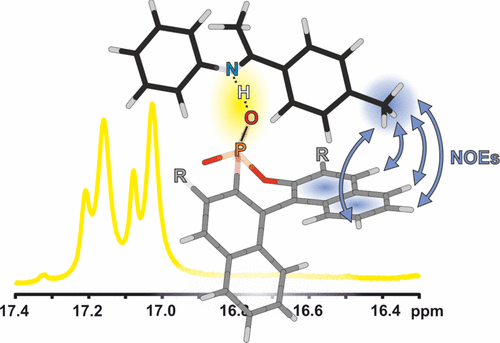
NMR Spectroscopic Characterization of Charge Assisted Strong Hydrogen Bonds in Brønsted Acid Catalysis
N. Sorgenfrei, J. Hioe, J. Greindl, K. Rothermel, F. Morana, N. Lokesh, R.M. Gschwind, J. Am. Chem. Soc., 2016, 138 (50), 16345-16354.
Hydrogen bonding plays a crucial role in Brønsted acid catalysis. However, the hydrogen bond properties responsible for the activation of the substrate are still under debate. Here, we report an in depth study of the properties and geometries of the hydrogen bonds in (R)-TRIP imine complexes (TRIP: 3,3′-Bis(2,4,6-triisopropylphenyl)-1,1′-binaphthyl-2,2′-diylhydrogen phosphate). From NMR spectroscopic investigations 1H and 15N chemical shifts, a Steiner–Limbach correlation, a deuterium isotope effect as well as quantitative values of 1JNH, 2hJPH and 3hJPN were used to determine atomic distances (rOH, rNH, rNO) and geometry information. Calculations at SCS-MP2/CBS//TPSS-D3/def2-SVP-level of theory provided potential surfaces, atomic distances and angles. In addition, scalar coupling constants were computed at TPSS-D3/IGLO-III. The combined experimental and theoretical data reveal mainly ion pair complexes providing strong hydrogen bonds with an asymmetric single well potential. The geometries of the hydrogen bonds are not affected by varying the steric or electronic properties of the aromatic imines. Hence, the strong hydrogen bond reduces the degree of freedom of the substrate and acts as a structural anchor in the (R)-TRIP imine complex.
__________________________________________________________________________________________________
Brønsted acid catalysis: hydrogen bonding versus ion pairing in imine activation

Nikola Kastner-Pustet - 02.10.2025 12:00


Phone: 0941 943-4626
Fax: 0941 943-4617
Room: CH23.1.80
E-Mail