

![]()
Unprecedented Mechanism of an Organocatalytic Route to Conjugated Enynes with a Junction to Cyclic Nitronates
V. Streitferdt, M.H. Haindl, J. Hioe, F. Morana, P. Renzi, F. von Rekowski, A. Zimmermann, M. Nardi, K. Zeitler, R.M. Gschwind, Eur. J. Org. Chem., 2019, 2019, 328-337.
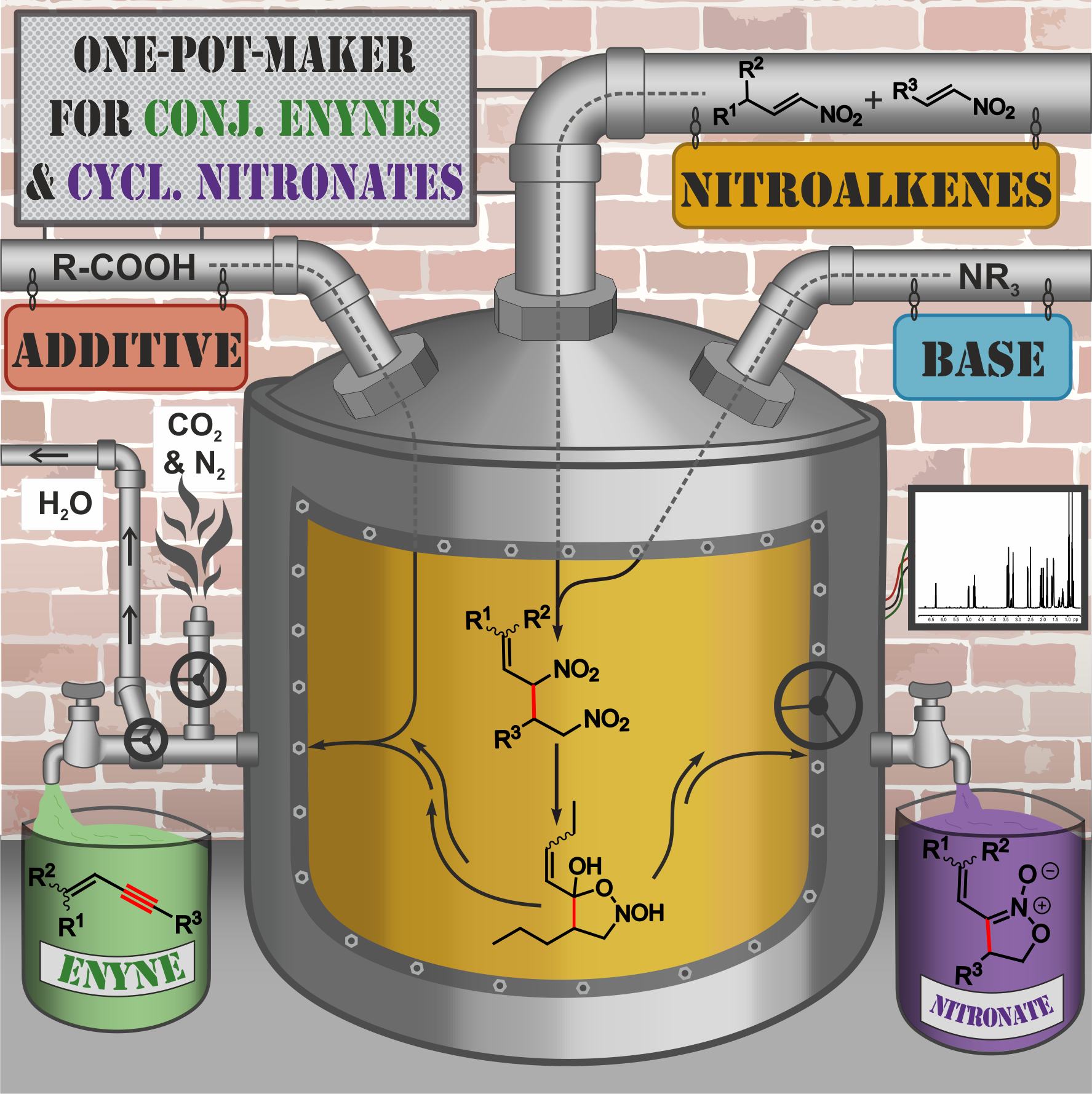
Conjugated enynes as well as cyclic nitronates are crucial building blocks for numerous natural products and pharmaceuticals. However, so far, no common and metal-free synthetic route to both conjugated enynes and cyclic nitronates has been reported. Herein, in situ NMR, labelling studies and theoretical calculations were combined to investigate the mechanism of the unusual triple bond formation towards conjugated enynes. Starting from nitroalkene dimers, first an isoxazolidine-2,5-diol derivative is formed as central intermediate. From this, enynes were generated by a combination of oxidation, dehydration, and retro 1,3-dipolar cycloaddition, whereas for nitronates a base induced intramolecular reorganization is proposed. While the product distribution could be controlled and high yields of nitronate were achieved, only medium to good yields for enynes were obtained due to polymerization losses. Nevertheless, we hope that these mechanistic investigations may provide a basis for further developments of organocatalytic or metal-free preparations of conjugated enynes and nitronates.
__________________________________________________________________________________________________
Chemical Exchange Saturation Transfer in Chemical Reactions: A Mechanistic Tool for NMR Detection and Characterization of Transient Intermediates
L. Nanjundappa, A. Seegerer, J. Hioe, R.M. Gschwind, J. Am. Chem. Soc., 2018, 140, 1855-1862.
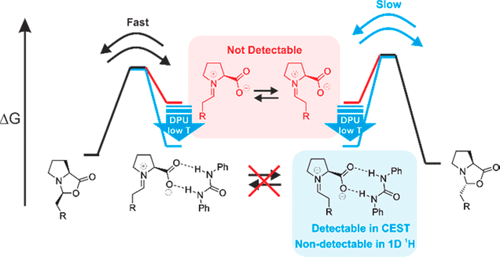
The low sensitivity of NMR and transient key intermediates below detection limit are the central problems studying reaction mechanisms by NMR. Sensitivity can be enhanced by hyperpolarization techniques such as dynamic nuclear polarization or the incorporation/interaction of special hyperpolarized molecules. However, all of these techniques require special equipment, are restricted to selective reactions, or undesirably influence the reaction pathways. Here, we apply the chemical exchange saturation transfer (CEST) technique for the first time to NMR detect and characterize previously unobserved transient reaction intermediates in organocatalysis. The higher sensitivity of CEST and chemical equilibria present in the reaction pathway are exploited to access population and kinetics information on low populated intermediates. The potential of the method is demonstrated on the proline-catalyzed enamine formation for unprecedented in situ detection of a DPU stabilized zwitterionic iminium species, the elusive key intermediate between enamine and oxazolidinones. The quantitative analysis of CEST data at 250 K revealed the population ratio of [Z-iminium]/[exo-oxazolidinone] 0.02, relative free energy +8.1 kJ/mol (calculated +7.3 kJ/mol), and free energy barrier of +45.9 kJ/mol (ΔG?calc.(268 K) = +42.2 kJ/mol) for Z-iminium → exo-oxazolidinone. The findings underpin the iminium ion participation in enamine formation pathway corroborating our earlier theoretical prediction and help in better understanding. The reliability of CEST is validated using 1D EXSY-build-up techniques at low temperature (213 K). The CEST method thus serves as a new tool for mechanistic investigations in organocatalysis to access key information, such as chemical shifts, populations, and reaction kinetics of intermediates below the standard NMR detection limit.
__________________________________________________________________________________________________
Enamine/Dienamine and Brønsted Acid Catalysis: Elusive Intermediates, Reaction Mechanisms, and Stereoinduction Modes Based on in Situ NMR Spectroscopy and Computational Studies
P. Renzi, J. Hioe, R.M. Gschwind
Over the years, the field of enantioselective organocatalysis has seen unparalleled growth in the development of novel synthetic applications with respect to mechanistic investigations. Reaction optimization appeared to be rather empirical than rational. This offset between synthetic development and mechanistic understanding was and is generally due to the difficulties in detecting reactive intermediates and the inability to experimentally evaluate transition states. Thus, the first key point for mechanistic studies is detecting elusive intermediates and characterizing them in terms of their structure, stability, formation pathways, and kinetic properties. The second key point is evaluating the importance of these intermediates and their properties in the transition state.
__________________________________________________________________________________________________
Remote-Stereocontrol in Dienamine Catalysis: Z-Dienamine Preferences and Electrophile–Catalyst Interaction Revealed by NMR and Computational Studies
A. Seegerer, J. Hioe, M. Hammer, F. Morana, P. Fuchs, R.M. Gschwind, J. Am. Chem. Soc., 2016, 138 (31), 9864-9873.
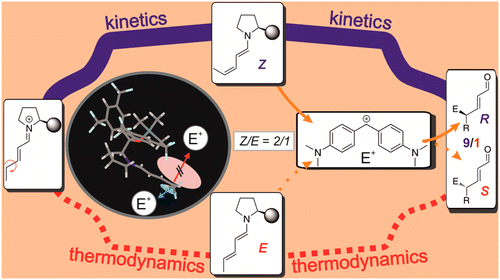
Catalysis with remote-stereocontrol provides special challenges in design and comprehension. One famous example is the dienamine catalysis, for which high ee values are reported despite insufficient shielding of the second double bond. Especially for dienamines with variable Z/E-ratios of the second double bond, no correlations to the ee values are found. Therefore, the structures, thermodynamics, and kinetics of dienamine intermediates in SN-type reactions are investigated. The NMR studies show that the preferred dienamine conformation provides an effective shielding if large electrophiles are used. Calculations at SCS-MP2/CBS-level of theory and experimental data of the dienamine formation show kinetic preference for the Z-isomer of the second double bond and a slow isomerization toward the thermodynamically preferred E-isomer. Modulations of the rate-determining step, by variation of the concentration of the electrophile, allow the conversion of dienamines to be observed. With electrophiles, a faster reaction of Z- than of E-isomers is observed experimentally. Calculations corroborate these results by correlating ee values of three catalysts with the kinetics of the electrophilic attack and reveal the significance of CH−π and stacking interactions in the transition states. Thus, for the first time a comprehensive understanding of the remote stereocontrol in γ-functionalization reactions of dienamines and an explanation to the “Z/E-dilemma” are presented. The combination of bulky catalyst subsystems and large electrophiles provides a shielding of one face and causes different reactivities of E/Z-dienamines in nucleophilic attacks from the other face. Kinetic preferences for the formation of Z-dienamines and their unfavorable thermodynamics support high ee values.
__________________________________________________________________________________________________
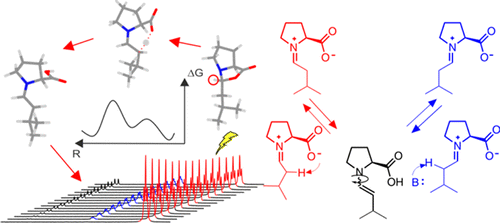
The Proline Enamine Formation Pathway Revisited in Dimethyl Sulfoxide: Rate Constants Determined via NMR
M. H. Haindl, J. Hioe, R. M. Gschwind, J. Am. Chem. Soc., 2015, 137 (40), 12835–12842.
Enamine catalysis is a fundamental activation mode in organocatalysis and can be successfully combined with other catalytic methods, e.g., photocatalysis. Recently, the elusive enamine intermediates were detected, and their stabilization modes were revealed. However, the formation pathway of this central organocatalytic intermediate is still a matter of dispute, and several mechanisms involving iminium and/or oxazolidinone are proposed. Here, the first experimentally determined rate constants and rates of enamine formation are presented using 1D selective exchange spectroscopy (EXSY) buildup curves and initial rate approximation. The trends of the enamine formation rates from exo-oxazolidinones and endo-oxazolidinones upon variation of the proline and water concentrations as well as the nucelophilic/basic properties of additives are investigated together with isomerization rates of the oxazolidinones. These first kinetic data of enamine formations in combination with theoretical calculations reveal the deprotonation of iminium intermediates as the dominant pathway in dimethyl sulfoxide (DMSO). The dominant enamine formation pathway varies according to the experimental conditions, e.g., the presence and strength of basic additives. The enamine formation is zero-order in proline and oxazolidinones, which excludes the direct deprotonation of oxazolidinones via E2 mechanism. The nucleophilicity of the additives influences only the isomerization rates of the oxazolidinones and not the enamine formation rates, which excludes a nucleophile-assisted anti elimination of oxazolidinones as a major enamine formation pathway.
__________________________________________________________________________________________________
What is your actual catalyst? TMS cleavage rates of diarylprolinol silyl ethers studied by in situ NMR

__________________________________________________________________________________________________
Stabilization of Proline Enamine Carboxylates by Amine Bases

__________________________________________________________________________________________________
Distinct conformational preferences of prolinol and prolinol ether enamines in solution revealed by NMR
Enamines, which are key intermediates in organocatalysis derived from aldehydes and prolinol or Jørgensen–Hayashi-type prolinol ether catalysts, were investigated conformationally in different solvents by means of NMR spectroscopy, in order to provide an experimental basis for a better understanding of the origin of stereoselection. For all of the enamines studied, surprisingly strong conformational preferences were observed. The enamines of the diarylprolinol (ether) catalysts were found to exclusively exist in the s-trans conformation due to the bulkiness of the pyrrolidine α-substituent. For prolinol enamines, however, a partial population of the s-cis conformation in solution was also evidenced for the first time. In addition, for all of the enamines studied, the pyrrolidine ring was found to adopt the down conformation. Concerning the exocyclic C–C bond, the sc-exo conformation, stabilized by CH/π interactions, is exclusively observed in the case of diarylprolinol ether enamines. In contrast, diarylprolinol enamines adopt the sc-endo conformation, allowing for an OH?N hydrogen bond and a CH/π interaction. A rapid screening approach for the different conformational enamine features is presented and this was applied to show their generality for various catalysts, aldehydes and solvents. Thus, by unexpectedly revealing the pronounced conformational preferences of prolinol and prolinol ether enamines in solution, our study provides the first experimental basis for discussing the previously controversial issues of s-cis/s-trans and sc-endo/sc-exo conformations. Moreover, our findings are in striking agreement with the experimental results from synthetic organic chemistry. They are therefore expected to also have a significant impact on future theoretical calculations and synthetic optimization of asymmetric prolinol (ether) enamine catalysis.

__________________________________________________________________________________________________
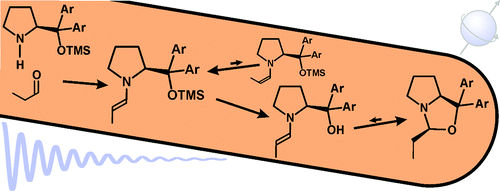
Formation and Stability of Prolinol and Prolinol Ether Enamines by NMR: Delicate Selectivity and Reactivity Balances and Parasitic Equilibria
M. B. Schmid, K. Zeitler,R. M. Gschwind, J. Am. Chem. Soc., 2011, 133 (18), 7065-7074.
Enamine key intermediates in organocatalysis, derived from aldehydes and prolinol or Jørgensen−Hayashi-type prolinol ether catalysts, were generated in different solvents and investigated by NMR spectroscopy. Depending on the catalyst structure, trends for their formation and amounts are elucidated. For prolinol catalysts, the first enamine detection in situ is presented and the rapid cyclization of the enamine to the oxazolidine (“parasitic equilibrium”) is monitored. In the case of diphenylprolinol, this equilibrium is fully shifted to the endo-oxazolidine (“dead end”) by the two geminal phenyl rings, most probably because of the Thorpe−Ingold effect. With bulkier and electron-withdrawing aryl rings, however, the enamine is stabilized relative to the oxazolidine, allowing for the parallel detection of the enamine and the oxazolidine. In the case of prolinol ethers, the enamine amounts decrease with increasing sizes of the aryl meta-substituents and the O-protecting group. In addition, for small aldehyde alkyl chains, Z-configured enamines are observed for the first time in solution. Prolinol silyl ether enamines are evidenced to undergo slow desilylation and subsequent rapid oxazolidine formation in DMSO. For unfortunate combinations of aldehydes, catalysts, solvents, and additives, the enamine formation is drastically decelerated but can be screened for by a rapid and facile NMR approach. Altogether, especially by clarifying the delicate balances of catalyst selectivity and reactivity, our NMR spectroscopic findings can be expected to substantially aid synthetically working organic chemists in the optimization of organocatalytic reaction conditions and of prolinol (ether) substitution patterns for enamine catalysis.
__________________________________________________________________________________________________
The Elusive Enamine Intermediate in Proline-Catalyzed Aldol Reactions: NMR Detection, Formation Pathway, and Stabilization Trends
The elusive enamine intermediate of nucleophilic proline catalysis was detected and stereochemically characterized by NMR analysis of the aldehyde self-aldolization reaction in dipolar aprotic solvents. NMR exchange spectroscopy (EXSY) was used to observe direct enamine formation from oxazolidinones. Additionally, the stabilization of the intermediate by the appropriate choice of solvent and substitution pattern on the aldehyde is presented.

Nikola Kastner-Pustet - 16.03.2023 09:28


Phone: 0941 943-4626
Fax: 0941 943-4617
Room: CH23.1.80
E-Mail