

![]()
Internal Acidity Scale and Reactivity Evaluation of Chiral Phosphoric Acids with different 3,3'-Substituents in Brønsted Acid Catalysis
K. Rel, M. Melikian, J. Hioe, J. Greindl, J. Gramüller, M.abka, N. Sorgenfrei, T. Hausler, F. Morana. R.M. Gschwind, Chem. Sci. 2019
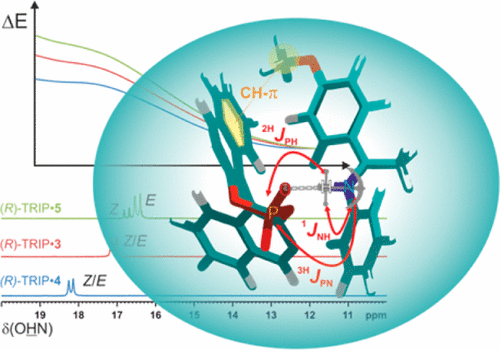
The concept of hydrogen bonding for enhancing substrate binding and controlling selectivity and reactivity is central in catalysis. However, the properties of these key hydrogen bonds and their catalyst-dependent variations are extremely difficult to determine directly by experiments. Here, for the first time the hydrogen bond properties of a whole series of BINOL-derived chiral phosphoric acid (CPA) catalysts in their substrate complexes with various imines were investigated to derive the influence of different 3,30-substituents on the acidity and reactivity. NMR 1H and 15N chemical shifts and 1JNH coupling constants of these hydrogen bonds were used to establish an internal acidity scale corroborated by calculations.
Deviations from calculated external acidities reveal the importance of intermolecular interactions for this key feature of CPAs. For CPAs with similarly sized binding pockets, a correlation of reactivity and hydrogen bond strengths of the catalyst was found. A catalyst with a very small binding pocket showed significantly reduced reactivities. Therefore, NMR isomerization kinetics, population and chemical shift analyses of binary and ternary complexes as well as reaction kinetics were performed to address the steps of the transfer hydrogenation influencing the overall reaction rate. The results of CPAs with different 3,30-substituents show a delicate balance between the isomerization and the ternary complex formation to be rate-determining. For CPAs with an identical acidic motif and similar sterics, reactivity and internal acidity correlated inversely. In cases where higher sterical demand within the binary complex hinders the binding of the second substrate, the correlation between acidity and reactivity breaks down.

__________________________________________________________________________________________________
NMR Spectroscopic Characterization of Charge Assisted Strong Hydrogen Bonds in Brønsted Acid Catalysis
N. Sorgenfrei, J. Hioe, J. Greindl, K. Rothermel, F. Morana, N. Lokesh, R.M. Gschwind, J. Am. Chem. Soc., 2016, 138 (50), 16345-16354.
Hydrogen bonding plays a crucial role in Brønsted acid catalysis. However, the hydrogen bond properties responsible for the activation of the substrate are still under debate. Here, we report an in depth study of the properties and geometries of the hydrogen bonds in (R)-TRIP imine complexes (TRIP: 3,3′-Bis(2,4,6-triisopropylphenyl)-1,1′-binaphthyl-2,2′-diylhydrogen phosphate). From NMR spectroscopic investigations 1H and 15N chemical shifts, a Steiner–Limbach correlation, a deuterium isotope effect as well as quantitative values of 1JNH, 2hJPH and 3hJPN were used to determine atomic distances (rOH, rNH, rNO) and geometry information. Calculations at SCS-MP2/CBS//TPSS-D3/def2-SVP-level of theory provided potential surfaces, atomic distances and angles. In addition, scalar coupling constants were computed at TPSS-D3/IGLO-III. The combined experimental and theoretical data reveal mainly ion pair complexes providing strong hydrogen bonds with an asymmetric single well potential. The geometries of the hydrogen bonds are not affected by varying the steric or electronic properties of the aromatic imines. Hence, the strong hydrogen bond reduces the degree of freedom of the substrate and acts as a structural anchor in the (R)-TRIP imine complex.
__________________________________________________________________________________________________
Brønsted acid catalysis: hydrogen bonding versus ion pairing in imine activation

__________________________________________________________________________________________________
Conformations, Conformational Preferences, and Conformational Exchange of N′-Substituted N-Acylguanidines: Intermolecular Interactions Hold the Key
R. Kleinmaier, M. Keller, P. Igel, A. Buschauer, R. M. Gschwind, J. Am. Chem. Soc., 2010, 132 (32), 11223-11233.
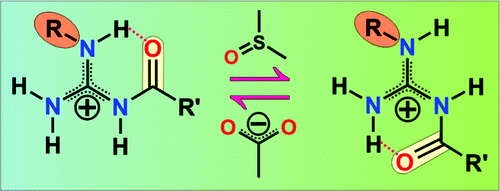
Guanidine and acylguanidine groups are crucial structural features of numerous biologically active compounds. Depending on the biological target, acylguanidines may be considered as considerably less basic bioisosteres of guanidines with improved pharmacokinetics and pharmacodynamics, as recently reported for N′-monoalkylated N-acylguanidines as ligands of G-protein-coupled receptors (GPCRs). The molecular basis for enhanced ligand−receptor interactions of acylguanidines is far from being understood. So far, only a few and contradictory results about their conformational preferences have been reported. In this study, the conformations, conformational preferences, and conformational exchange of four unprotonated and seven protonated monoalkylated acylguanidines with up to six anions and with bisphosphonate tweezers are investigated by NMR. Furthermore, the effects of the acceptor properties in acylguanidine salts, of microsolvation by dimethylsulfoxide, and of varying acyl and alkyl substituents are studied. Throughout the whole study, exclusively two out of eight possible acylguanidine conformations were detected, independent of the compound, the anion, or the solvent used. For the first time, it is shown that the strength and number of intermolecular interactions with anions, solvent molecules, or biomimetic receptors decide the conformational preferences and exchange rates. One recently presented and two new crystal structures resemble the conformational preferences observed in solution. Thus, consistent conformational trends are found throughout the structurally diverse compound pool, including two potent GPCR ligands, different anions, and receptors. The presented results may contribute to a better understanding of the mechanism of action at the molecular level and to the prediction and rational design of these biologically active compounds.
__________________________________________________________________________________________________
The H-Bonding Network of Acylguanidine Complexes: Combined Intermolecular 2hJH,P and 3hJN,P Scalar Couplings Provide an Insight into the Geometric Arrangement
G. Federwisch, R. Kleinmaier, D.Drettwan, R. M. Gschwind, J. Am. Chem. Soc., 2008, 130 (50), 16846-16847.
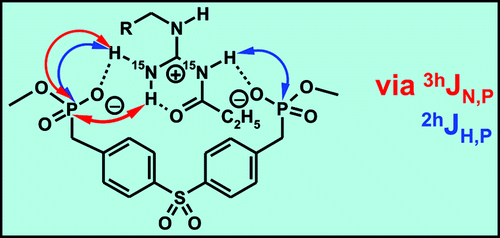
The H-bonding networks of arginines and acylguanidines are crucial for many biological and pharmaceutical interactions. However, the effect of acylation of guanidines on the binding mode and the H-bond strengths has not yet been explored in solution. Therefore, the H-bonding network of a 15N labeled acylguanidine derivative in a bisphosphonate arginine receptor is investigated. The direct NMR detection of 1D, 2D, and 3D correlations caused by 2hJH,P and, for the first time in nonbiomacromolecules, 3hJN,P couplings, allows for the geometric analysis of the H-bonding network and indicates an end-on binding mode with two different POH angles. The acylguanidine adopts the same binding mode as the corresponding guanidine but forms significantly stronger H-bonds. This may explain the success of acylguanidine ligands in medicinal chemistry applications.
__________________________________________________________________________________________________
NMR Detection of Intermolecular NH···OP Hydrogen Bonds between Guanidinium Protons and Bisposphonate Moieties in an Artificial Arginine Receptor
R. M. Gschwind, M. Armbrüster,I. Z. Zubrzycki, J. Am. Chem. Soc., 2004, 126 (33), 10228-10229.
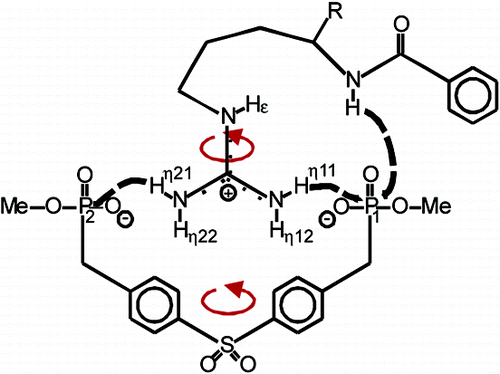
Hydrogen bonding plays a major role in the selective recognition of guanidinium groups by receptor molecules. The present NMR investigation provides direct experimental evidence of hydrogen bonds in an artificial arginine receptor complex consisting of α-N-benzoylarginine ethyl ester and a bisphosphonate tweezers molecule. trans-Hydrogen bond 2hJHP couplings between the phosphonate moieties and individual guanidinium protons as well as the amide proton have been detected by [1H,31P]-HMBC and [31P,1H]-INEPT experiments. The detected hydrogen bonding network in the investigated artificial arginine receptor shows a symmetrical end-on interaction of the guanidinium moiety, which enables concerted rotations and deviates from the structure proposed for the biological arginine fork.
Nikola Kastner-Pustet - 16.03.2023 09:27


Phone: 0941 943-4626
Fax: 0941 943-4617
Room: CH23.1.80
E-Mail